
_Gail Rosen
Rosen is a professor in the Department of Electrical and Computer Engineering in the College of Engineering. She heads Drexel's Center for Biological Discovery from Big Data.
Colonies of bacteria and viruses naturally coexist throughout the human body and play roles in digestion, metabolism and even fighting off diseases. But for scientists, understanding just how they do it remains a question.
Researchers are hoping to find answers by treating the human biome as a “big data” problem. They’re using pattern-recognition algorithms and machine learning to sift through massive amounts of genetic sequencing information that has come available in recent years. Their goal is to identify groupings of microbial communities that occur in concert with each other.
Meet_Metagenomics
Metagenomics is a field of science that applies a computational approach to studying organism interactions and evolution. In this type of research, a scan of a genetic material sample — DNA or RNA — can be interpreted to reveal the organisms that are likely present. The method presented by Rosen’s group takes that one step further by analyzing the genetic code to spot recurring patterns, an indication that certain microbes present are found together so frequently that it’s not a coincidence. Rosen’s team calls this “themetagenomics,” because they are looking for recurring themes in microbiomes that are indicators of co-occurring groups of microbes..
Their findings, published in PLOS ONE, put forth a new method of analyzing the codes found in microbial RNA to reveal how these communities operate.
“There are thousands of species of microbes living in the body, so if you think about all the permutations of groupings that could exist you can imagine what a daunting task it is to determine which of them are living in community with each other,” says Gail Rosen, who co-authored the paper with Steve Woloszynek, an MD-PhD trainee in the College of Medicine. “Our method puts a pattern-spotting algorithm to work on the task, which saves a tremendous amount of time and eliminates some guesswork.”
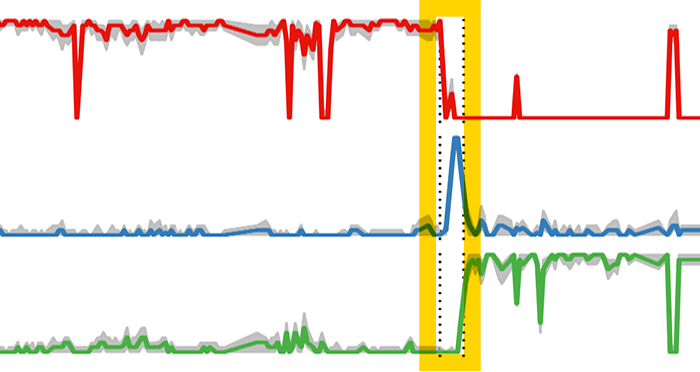
gut_reaction
The blue line represents the probability of a Salmonella infection (suspected from bad sushi) in a subjects’ gut. Researchers observed a drastic change in the microbial composition of the person’s gut before and after infection (the yellow bar). This change is signified by the probablility of the pre-infection microbiome in red, which declines and is replaced by the probability of the green line. .
Current methods for studying microbiota, gut bacteria for example, take a sample from an area of the body and then look at the genetic material that’s present. This process inherently lacks important context, according to the authors.
“Most metagenomics methods just tell you which microbes are abundant, but they don’t really tell you much about how each species is supporting other community members,” Rosen says. “With our method you get a picture of the configuration of the community — for example, it may have E. coli and B. fragilis as the most abundant microbes and in pretty equal numbers — which may indicate that they’re cross-feeding. Another community may have B. fragilis as the most abundant microbe, with many other microbes in equal, but lower, numbers — which could indicate that they are feeding off whatever B. fragilis is making, without any cooperation.”